
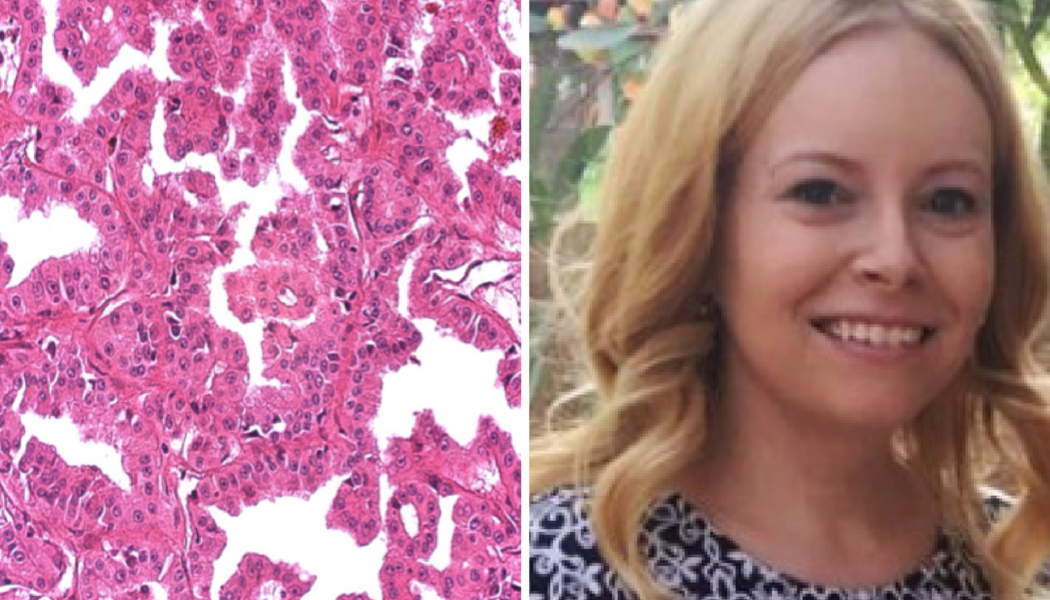
Laura Esfeller was diagnosed with stage 4 type 2 papillary kidney cancer when she was only 29 years old. She would later learn that she also has a rare genetic condition called Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC), also known as Reed’s Syndrome. This is her story.
Kidney cancer is a rare enough disease, but what happens when you have a rare subset of a rare disease?
I was diagnosed with stage IV type 2 papillary renal cell carcinoma a few days after I turned 29 years old. It was a shock, to say the least, and like most cancer patients, I asked myself and my doctors, “Why? Why me?”
I didn’t know it at the time, but I have a rare genetic condition called Hereditary Leiomyomatosis and Renal Cell Cancer, also known as Reed’s Syndrome. Since it’s a mouthful, most people call it HLRCC. ASCO’s Cancer.net describes it as “a hereditary condition associated with multiple leiomyomas, which are smooth muscle tumors arising in the skin, uterine fibroids, which are non-cancerous growths in a woman’s uterus, and type 2 papillary renal (kidney) cancer.”
Although doctors have been researching it for decades, HLRCC was only officially named a disease in 2001. It’s caused by a mutation in one copy of the fumarate hydratase (FH) gene. It increases a person’s risk of kidney cancer by as much as 30%. Less than 1000 families across the world are known to carry this mutation, but it is believed that many more have it but are unaware due to this being such a new discovery.
Not everyone who has HLRCC develops skin tumors, much less kidney cancer. Most people who do get these leiomyomas begin developing them in their 20s. They can be painful but they are benign. I began developing leiomyomas on my back about 2 years before I was diagnosed with kidney cancer. I’ve had a few more develop since then. Luckily mine are not painful. I’ve also had uterine fibroids since my early 20s. Unfortunately, since leiomyomas are benign and fibroids are common in women, neither was alarming to my doctors.
My story isn’t uncommon among HLRCC patients. Although the average kidney cancer patient is in their 60s upon diagnosis, many HLRCC patients who develop kidney cancer are diagnosed with it anywhere from their 20s to 40s, although younger and older patients are not unheard of. Sadly, because this condition is so unheard of, many who do develop kidney cancer are also like me in that they are stage IV upon diagnosis. HLRCC is also typically more aggressive and difficult to treat.
The unusualness of this disease makes it even more imperative for patients to see a specialist immediately, and for anyone who is diagnosed with renal cell carcinoma, even if it is not papillary type II, under the age of 45 to undergo genetic testing. Those who have HLRCC but do not have kidney cancer are advised to undergo yearly abdominal/pelvic CT scans or a MRI each year.
I was fortunate to be admitted to a clinical trial for papillary kidney cancer patients in 2016 after my radical nephrectomy, and I am now NED (no visible evidence of disease) on Cabometyx. I know how incredibly lucky I am to have had such amazing results on targeted therapy, not to mention my world class oncologist, and I don’t take it for granted. I believe it is crucial for patients to educate themselves about their disease and treatment options, and to enroll in clinical trials when possible. Clinical trials provide scientists the opportunity to treat rare diseases they may not otherwise encounter and allow access to cutting-edge treatments, not to mention paving the way for future patients. It is because patients before me were brave enough to try a clinical trial that I can live my life again, and I am eternally grateful for their courage.
For more information on HLRCC you can find additional information through the HLRCC Family Alliance and DrivenToCure.org.
Laura Esfeller serves on the KCCure Patient Advisory Board and is part of our papillary kidney cancer community.







